
This AI Act Medical Devices Expert Perspective examines why many industry professionals believe medical AI was already heavily regulated before the EU AI Act.
This article is part of our EU AI Act Learning Guide, where you can find all four articles in this series.
Personal note from the author
I make no apology for saying this plainly: the inclusion of medical devices in the EU AI Act is one of the most damaging regulatory mistakes Europe has made in the field of medical AI.
Three years ago, I argued directly and repeatedly with those that informed the EU Parliament, that regulated medical devices should not be swept into the EU AI Act as if they were ordinary software tools, social-scoring systems, recruitment algorithms, or consumer-facing AI products.
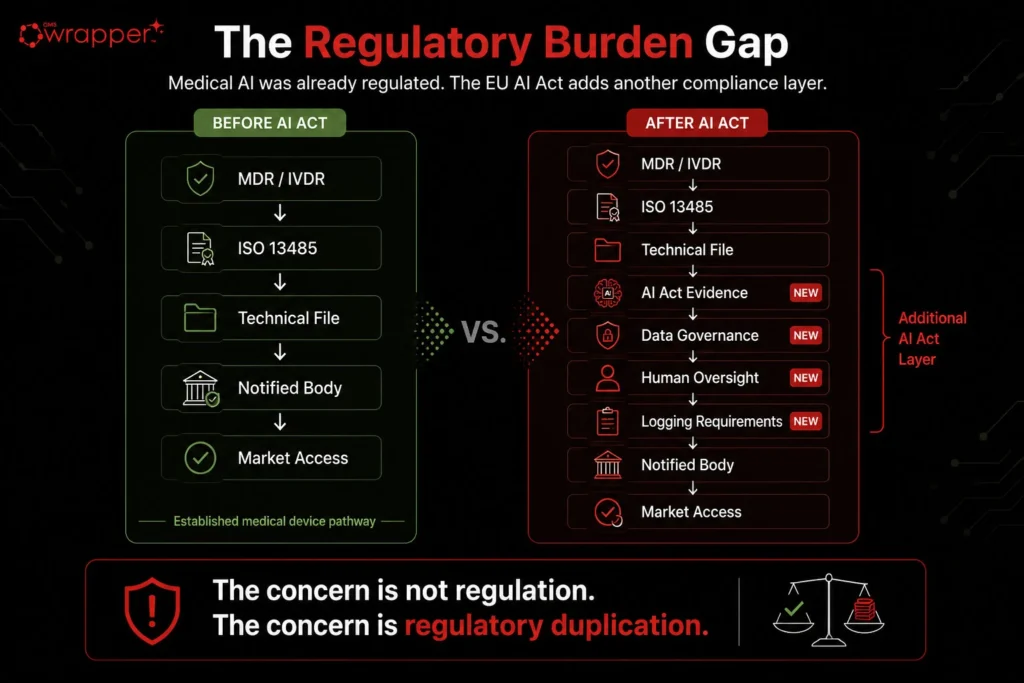
Medical devices were never an unregulated frontier.
AI used in a medical device is already subject to clinical validation, risk management, usability controls, cybersecurity review, technical documentation, post-market surveillance, notified-body scrutiny and continuous quality-system obligations under the existing medical-device framework. Anyone who has actually built, validated, certified, and defended a medical AI device understands this.

That distinction was ignored.
In my view, the inclusion of medical devices in the EU AI Act was not driven by a demonstrated safety gap. It was ramp driven regulatory overreach, political momentum, and a profound, ego induced, solipsistic blindness to what medical-device validation already requires – understanding it didn’t serve their ends. The result is not smarter, safer regulation.
In my view, the inclusion of medical devices in the EU AI Act was not driven by a demonstrated patient-safety gap. It was driven by regulatory overreach, political momentum, and a profound refusal to understand what medical-device validation already requires. Medical AI was never the Wild West. It was already governed by clinical evaluation, risk management, technical documentation, post-market surveillance, notified-body review, and quality-system control. But that truth was inconvenient, because it did not serve the expansion of the AI Act. So the truth was pushed aside. The result is not smarter regulation. It is not safer regulation. It is regulatory duplication imposed as virtue, control dressed as protection, hand-cuffing European innovation in the face of American competition.
This was regulatory vanity, masquerading as patient safety.
Europe had an opportunity to create a sophisticated innovation driven carve-out: keep medical AI under MDR/IVDR, strengthen guidance where needed, and place it in a lighter, sector-integrated framework comparable to the treatment available to certain other already-regulated product sectors. Instead, medical AI was pulled into a horizontal AI law designed largely for very different risks.
That decision matters.
It adds cost, delay, uncertainty and legal exposure to the very companies Europe claims it wants to support. It burdens SMEs and innovators while favouring larger US companies with deeper compliance teams, larger legal budgets and more patience for bureaucratic friction. In practical terms, Europe has handed American medical AI companies an advantage they did not earn. It was a European self-inflicted wound.
This was not a victory for patient safety. It was a failure of regulatory intelligence.
And the idea that medical devices may later be moved into a more appropriate annex B or lighter framework is, frankly, naïve. Bureaucracies never surrender control once they have acquired it. Once a sector is captured by a new regulatory regime, the default direction is not simplification. It is expansion. Rules for rules sake.
That is the real tragedy of the EU AI Act for medical devices: it did not address patient safety, it did not solve an identified clinical problem.
It was regulatory overreach dressed up as public protection.
I have no apology for my opinion, I only regret my voice was not louder.
What qmsWrapper is!
qmsWrapper™ is an end-to-end Event-to-Evidence AI Quality Management and Compliance Platform built for medical-device startups and SMEs. Designed to support companies navigating the complex realities of MDR, FDA, ISO 13485, and emerging EU AI Act requirements, qmsWrapper™ provides a central, controlled repository for the critical documentation, records, and evidence required by regulators, notified bodies, customers, and auditors.
From EUDAMED obligations and technical-file maintenance to PMS, vigilance, audit preparation, and EU AI Act evidence, qmsWrapper™ goes beyond traditional document management. It uses AI to help connect day-to-day company events directly to the regulatory evidence they create or affect, while keeping the human in control of all regulatory decisions and actions. The result is a visible, traceable, and audit-ready quality ecosystem.
The platform is purpose-built to scale with a company’s growth. Whether a startup is moving away from paper forms, spreadsheets, and shared drives, or an SME managing advanced quality and regulatory workflows, qmsWrapper™ provides a practical step-by-step path: establish controlled processes and forms, scale into a connected QMS, and then expand into vigilance, regulatory evidence, and audit-readiness.
To make this progression manageable, qmsWrapper™ is structured into three distinct layers:
Wrapper Process establishes the foundation with controlled digital forms, taskforms, logs, processes, and guided ISO 13485 workflows.
Wrapper QMS connects core quality events, including CAPA, change control, deviations, non-conformities, supplier management, training, and risk-linked workflows.
Wrapper Vigilance supports advanced PMS, clinical evidence, technical-file maintenance, EUDAMED preparation, EU AI Act evidence, vigilance reporting, regulatory traceability, and AI-assisted audit preparation.
Ultimately, qmsWrapper™ supports medical-device innovation from early-stages of development through regulatory approval and long-term market access. It makes compliance visible, connected, traceable, and manageable, without pretending that regulation is simple.
Book a meeting with qmsWrapper.
Notwithstanding.
Notwithstanding the flaws and burdens created by the EU AI Act, qmsWrapper is taking a practical position: help startups and SMEs survive it. qmsWrapper cannot remove the regulatory burden, but it can make that burden more visible, more structured, and more manageable. Its AI functions are validated for their intended purpose and documented to support compliance with the EU AI Act: assisting users in preparing, organising, linking, and maintaining the evidence required for medical-device regulatory compliance. qmsWrapper does not replace the responsible person, the quality manager, the regulatory lead, or the manufacturer’s legal accountability. It makes clear where AI is used, keeps the human in control, and preserves the deterministic role of the user in every regulatory decision and action. In that sense, qmsWrapper does not pretend the EU AI Act is good law. It simply gives SMEs a way to comply with it without being crushed by it.
Because when the devil makes you dance, better to pick your own tune.
The debate about whether medical devices should have been included in the EU AI Act will likely continue for years. What is certain, however, is that manufacturers must now operate within both frameworks. For startups and SMEs, the challenge is no longer whether the regulation exists, but how to manage it efficiently.
Where Medical Device Companies Go From Here
Does the EU AI Act replace MDR or IVDR requirements?
No. Medical device manufacturers must still comply with MDR or IVDR requirements. The EU AI Act adds additional obligations for AI systems that fall within its scope.
Are AI medical devices already regulated without the EU AI Act?
Yes. AI medical devices are already subject to clinical evaluation, risk management, technical documentation, post-market surveillance, quality management system requirements, and notified body review under MDR, IVDR, ISO 13485, and related standards.
Why do some industry experts oppose the inclusion of medical devices in the EU AI Act?
Critics argue that medical AI was already heavily regulated and that the AI Act creates overlapping compliance requirements without addressing a clearly identified patient-safety gap.
Will the EU AI Act increase compliance costs for medical device SMEs?
Many industry observers believe it will. Additional documentation, governance, oversight, logging, and evidence requirements may increase the cost and complexity of bringing AI-enabled medical devices to market.
Can software help companies manage EU AI Act compliance?
Software cannot remove regulatory obligations, but it can help organize documentation, maintain traceability, manage evidence, and support audit readiness across MDR, ISO 13485, and EU AI Act requirements.


