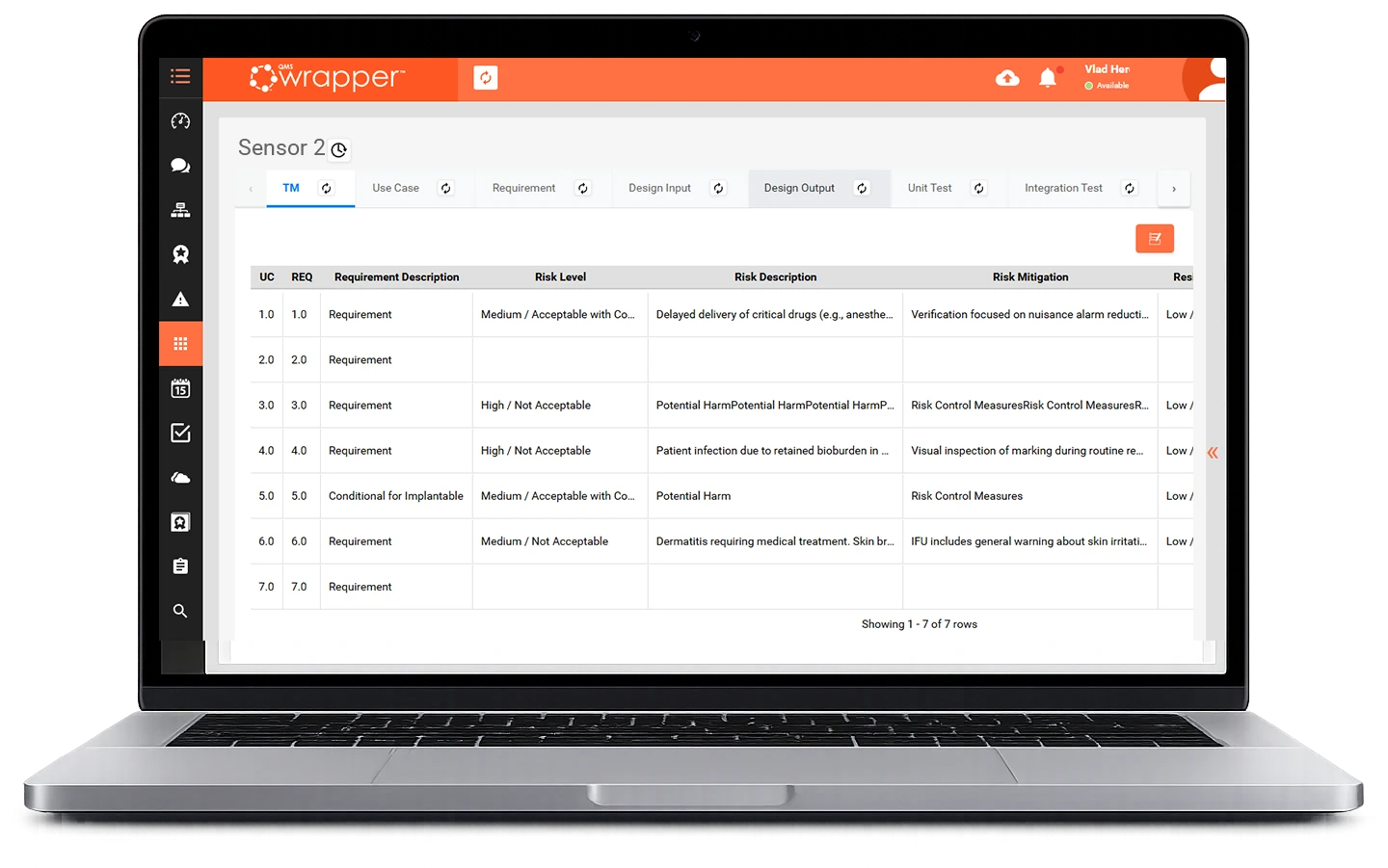
Design Control Traceability Matrix
Design Control That Survives Change
qmsWrapper provides a Design Control Traceability Matrix built to support structured documentation across the FDA Design History File (DHF) and EU MDR Technical File.
It connects user needs, requirements, risks, design inputs and outputs, verification, and validation into one maintained structure.
Linked records and controlled updates help preserve consistency as your design evolves.
When design changes occur, linked elements can be reviewed within the same structured environment.
Traceability Is Not a Table. It’s a System.
In many systems, the Traceability Matrix is treated as a generated report — reviewed primarily before audits.
In qmsWrapper, the Design Traceability File (DTF) is built on structured forms and linked records.
Each requirement, risk, test, and design output exists as an individual, revision-controlled record — connected through structured references.
The problem: Traceability Breaks When Things Change
When traceability depends on manual linking, spreadsheets, or exported reports, design control becomes difficult to maintain consistently.
Design control is tested when requirements evolve, risks are reassessed, or verification results require updates.
If links are maintained outside the system, consistency depends on manual effort rather than structure.
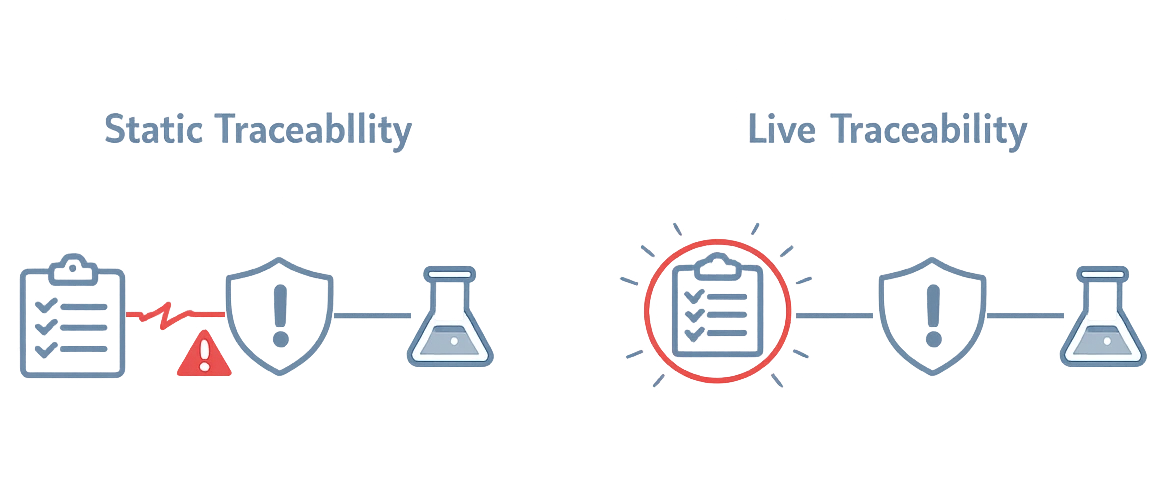
Traceability should follow the work, not chase it.
When Events Affect Design, Traceability Must Be Reviewed
Design changes rarely happen in isolation.
They may be triggered by Events like deviations, nonconformity, change or feedback.
In qmsWrapper, design-related records remain accessible within the same structured environment as quality records.
When a design element is updated, linked requirements, risks, and tests can be reviewed to ensure consistency.
The system supports structured review — helping teams maintain alignment across related records.
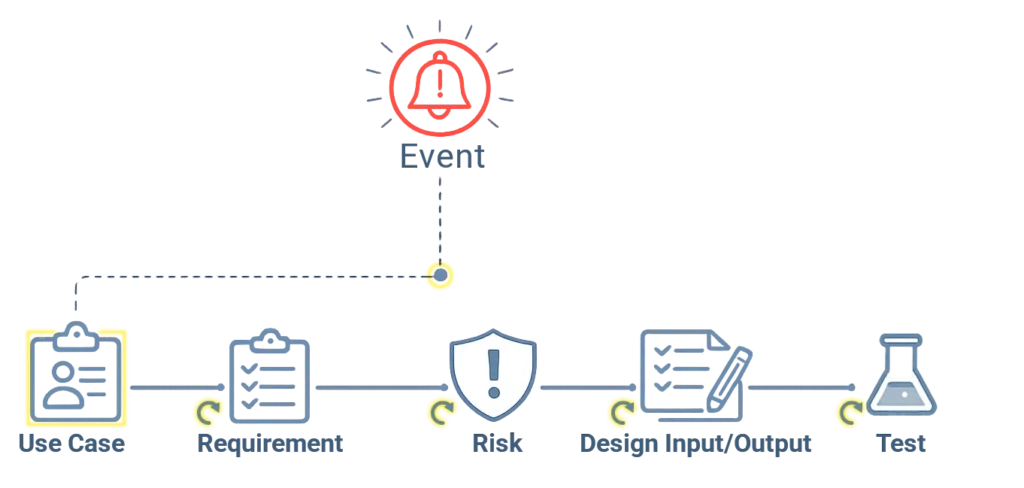
Every design decision should explain itself.
AI-Assisted Record Review
Managing design relationships can be complex.
qmsWrapper provides AI-assisted support for reviewing linked risks, locating related test protocols, and searching across connected records.
AI supports navigation and review.
Final design decisions remain fully controlled by the user.

Built for Regulatory Review
The Traceability Matrix supports:
- DHF and Technical File structure
- 510(k) submissions
- CE Marking under MDR
Auditors and Notified Bodies can review the traceability chain from requirement to evidence within a structured environment.
Clear record linkage reduces the need for manual reconstruction before audits.
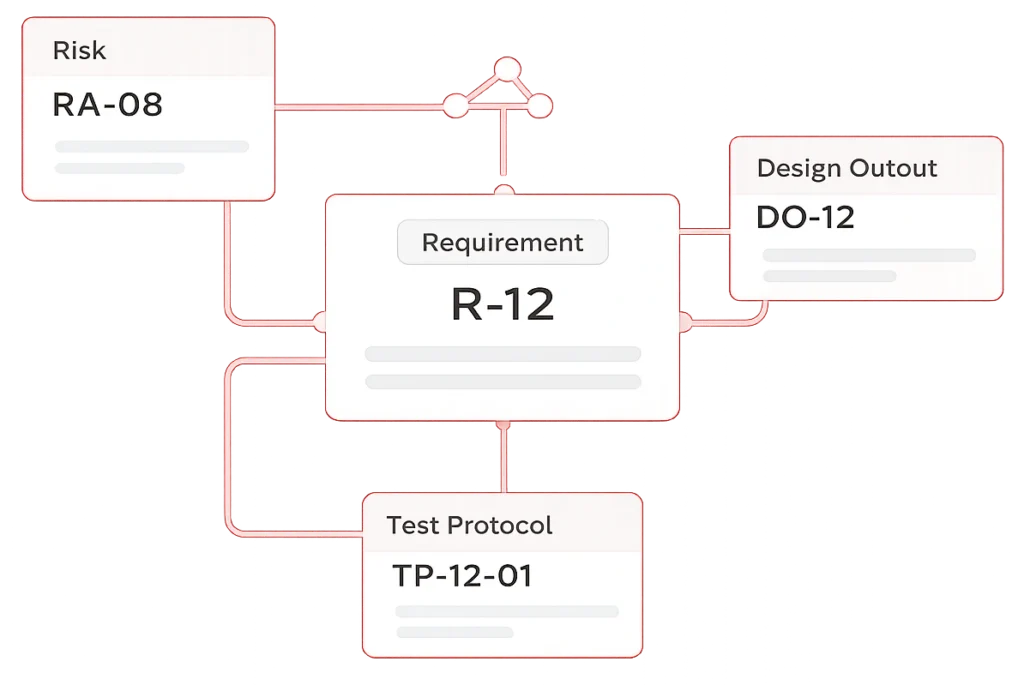
More Than a Matrix — A Structured Design Traceability File
The Design Traceability File (DTF) supports both the FDA Design History File (DHF) and the EU MDR Technical File within one structured documentation environment.
It preserves the logical relationship between requirements, risks, and evidence — ensuring consistency as your design evolves.
Regulators do not review isolated documents.
They review traceable design reasoning.
The DTF maintains that continuity.
“Traceability is evidence of design logic”
Design Control That Evolves With Your Device
qmsWrapper treats traceability as a system — not a deliverable.
If you are preparing for MDR, FDA 21 CFR 820, or ISO 13485 audits, a structured and consistently maintained traceability system can reduce reconstruction effort and audit risk.
Book a meeting to see how the Design Control Traceability Matrix supports your regulatory documentation process.
Frequently Asked Questions About Design Control and Traceability in Medical Devices
What is a Design Control Traceability Matrix in medical devices?
A Design Control Traceability Matrix in medical devices links user needs, design requirements, risks, design inputs and outputs, and verification and validation evidence within a structured documentation system. It ensures that every requirement is traceable to risk analysis and test results in accordance with FDA 21 CFR 820 and ISO 13485.
Is a Traceability Matrix required for FDA and MDR compliance?
While regulations do not mandate a specific “matrix” format, both FDA 21 CFR 820 and EU MDR require documented design control and traceability. A structured traceability matrix helps demonstrate that requirements, risks, and verification activities are consistently linked within the Design History File (DHF) and Technical File.
How does traceability support ISO 13485 design control?
ISO 13485 requires documented design inputs, outputs, review, verification, validation, and risk management. A Design Control Traceability Matrix supports compliance by maintaining structured relationships between these elements throughout the product lifecycle.
What is the difference between a Design History File and a Technical File?
The Design History File (DHF) is an FDA requirement documenting design control activities under 21 CFR 820. The Technical File under EU MDR contains similar evidence but follows European regulatory structure. A structured traceability system helps maintain consistency between both documentation sets.
How does traceability reduce audit risk?
Traceability reduces audit risk by ensuring that requirements, risks, and test evidence are linked within a controlled system. This minimizes manual reconstruction before inspections and allows auditors to follow the chain of design reasoning without relying on disconnected documents.
Can AI be used in design control documentation?
AI can assist with reviewing linked records, identifying related risks or test protocols, and retrieving documentation across modules. However, regulatory responsibility remains with the manufacturer, and final design decisions must remain under human control.